")
")

Récemment, le laboratoire où je travaille a publié un nouvel article. Comme le sujet est relativement compliqué, je me suis dit qu’au lieu de toujours essayer de réexpliquer oralement (ce qui n’est pas simple vu la longueur de l'explication dans laquelle je me lance), j’allais plutôt tenter de faire l’exercice de donner une explication que tout le monde peut comprendre sans avoir suivi des cours de biologie.
L’article que nous avons publié est un condensé en 8 pages de plusieurs années de recherche, c’est le fruit d'un long travail qui implique de nombreuses personnes et je suis fier d’avoir pu participer à ce projet avec tous les autres auteurs.
Je ne vais pas révéler le titre de l'article parce qu'il "spoil" un peu tous nos résultats intéressants (les articles scientifiques ne sont pas écrits comme des thrillers). Donc je vais essayer de rendre ça intéressant (bon courage!)...
On étudie Adineta vaga, un animal microscopique dont vous pouvez voir quelques images ci-dessous... (et que je vais directement commencer à appeler A. vaga car c'est l'usage scientifique et mais surtout par facilité).
Les bdelloides
Les photos suivantes sont des grossissements car ces animaux sont pratiquement invisibles à l’œil nu. A. vaga fait partie d’un groupe d’animaux peu connus, les rotifères bdelloïdes (à vos souhaits !).





Peu importe ces noms compliqués, ce n’est pas ce qui est intéressant. Mais du coup pourquoi A. vaga et plus généralement les rotifères bdelloïdes sont intéressants ?
Une première caractéristique intéressante est que les bdelloïdes survivent à des conditions extrêmes : ils sont capables de vivre dans l’eau et dans le désert, en antarctique. Mais ils sont encore plus résistants, ils sont capable de survivre à des doses complètement absurdes d'irradiations ionisantes (par exemple des doses de rayons X qui tueraient un être humain en quelques secondes).
Cependant, cette extrême résistance n'est pas leur seul point d'intérêt. Les premières observations de bdelloïdes ont eu lieu aux alentours de 1670 (déjà au 17ème siècle!). Et depuis qu'ils sont connus et observés, aucun mâle n'a été observé. Il n'y a que des femelles : ces animaux sont asexués. Et c'est assez facile à vérifier, on peut attraper un oeuf qui vient d'être pondu par une femelle et l'isoler dans une boîte de pétri. Il suffit alors d'attendre, quelques jours et on observe maintenant plusieurs rotifères dans la boîte!
L'asexualité n'est pas un phénomène rare, on trouve régulièrement des espèces asexuées. C'est le cas de beaucoup de plantes, mais aussi des bactéries, etc. Cependant, c'est plutôt rare chez les animaux multicellulaires.
Bon maintenant qu'on sait pourquoi ils sont intéressants, revoyons les bases de la biologie :)
Biology 101
Une première notion importante : les cellules. Tous les animaux, les plantes et les bactéries sont composées de cellules : des unités autonomes microscopiques qui sont capables de se diviser et de se spécialiser. Nous sommes des animaux et nous en avons plusieurs sortes : des cellules musculaires capables de se contracter, des neurones capables de conduire un signal électrique, des cellules de peau, des cellules intestinales, des cellules sécrétrices, des cellules sanguines (les globules), etc.
Ces cellules s’organisent en tissus comme les fibres musculaires, les nerfs, les glandes, la peau, le sang, etc. Et ces tissus s’agencent en organes comme le cerveau, le cœur, le pancréas, les vaisseaux sanguins, etc. Ces tissus peuvent même créer des matières « non-vivantes » comme les poils, la peau, les os.
J’adore la vidéo ci-dessous qui montre le développement d’un embryon de triton. Essayez de repérer sur la vidéo les moments où on passe d’un tas de cellules à un tissu, d’un tissu à un organe et d’un groupe d’organes à un organisme composé de milliards de cellules voire de milliers de milliards de cellules.
Ces moments ne sont pas clairement déterminés mais progressifs. C’est un continuum, comme énormément de phénomènes en biologie et en général.
Adineta vaga est beaucoup plus petit qu’une salamandre et les femelles adultes contiennent environ 1000 cellules. C’est vraiment beaucoup (beaucoup) moins qu’un être humain adulte qui compte approximativement trente-sept milles milliards cellules (37,000,000,000,000).
Plan de fabrication, génome, bibles et quelques remises à l’échelle…
Le plan qui permet à ces cellules de s’organiser s’appelle le génome. Le plan (le génome) humain, s’il était écrit avec des lettres sous forme d’une liste d’instruction, contiendrait une séquence de 6,4 milliards de lettres. Heureusement, environ la moitié de cette information est redondante : elle est présente en deux copies (2 versions) : une qui provient du père et une autre qui provient de la mère.
Donc si on résumait la quantité totale d’information non-redondante, on pourrait garder une seule des deux copies, on a donc une taille de génome à environ 3,2 milliards de lettres.
Pour vous donner une idée de ce que ce nombre représente en quantité totale d’information, j'ai compté le nombre de lettres dans une bible anglaise (version King James parce qu'elle est facile à obtenir sur internet). Elle contient environ 3 millions 600 milles caractères (sans compter les espaces). Il faudrait donc 889 bibles pour contenir une version non-redondante d’un génome humain.
Presque toutes les cellules d’un organisme contiennent le génome complet de l'organisme. C’est comme si tous les ouvriers d’un chantier se promenaient avec les instructions de fabrications du bâtiment entier (et que ces instructions sont écrites dans 890 équivalent-bibles).
Heureusement pour ces ouvriers, ils ne doivent pas lire toutes les instructions à chaque fois. En réalité, chaque cellule peut lire uniquement les parties importantes pour son travail. C’est comme si on avait donné un énorme plan dépliable complet aux ouvriers mais qu’on avait collé les pages qui ne concernent pas la fonction de l’ouvrier, le plombier ne peut lire que la partie importante à la plomberie du bâtiment, pareil pour le maçon, etc. Il y a aussi quelques ouvriers qui ne se spécialisent pas, ils sont un peu comme les formateurs. Le rôle des cellules non spécialisées est de se diviser (et pour ça il faut copier le plan) pour produire des cellules spécialisées avec un plan complet mais qui n’ont pas besoin de tout lire et donc on des pages collées. On appelle ce processus, la différentiation.
Évidemment, l’évolution n’a pas sélectionné des cellules qui lisent des plans ou des livres, cette façon de stocker l’information. Pour pouvoir stocker autant d’information dans un tout petit espace (quelques millionièmes de millimètre) et surtout pouvoir lire l’information en très peu de temps, les cellules utilisent des molécules d’ADN.
L’ADN est une énorme molécule composée principalement d’atomes de carbone (noté C), d’hydrogène (H), d’azote (N) et de phosphore (P). Ces éléments sont associés en unités (qu'on représente habituellement par des lettres) qu’on appelle les nucléotides. On distingue 4 nucléotides qui diffèrent légèrement par leur composition : A, T, C et G (Adénine, Thymine, Cytosine et Guanine respectivement).
L'ADN dans une cellule peut être plus ou moins condensé (c'est à dire que la molécule est plus ou moins repliée sur elle même d'une certaine façon). La forme la plus condensée est appelée le chromosome et la forme la moins condensée est la forme d'ADN la plus connue : la double hélice.
On parle de double hélice parce qu'en réalité, une molécule d'ADN est composée de deux brins chaque brin est une suite de nucléotide telle que ATTCGTTAGCTA...etc. Les brins sont complémentaires c'est à dire que pour chaque nucléotide sur un brin il y a un autre nucléotide complémentaire sur l'autre brin. A est complémentaire avec T et C avec G. En pratique si on connait n'importe quel brin, on connaît automatiquement l'autre brin il suffit de convertir les A en T et les C en G et réciproquement pour T et G.
On mesure la taille des séquences parfois en nucléotides (nt) mais plus fréquemment, on utilise une unité appelée la "paire de bases" (écrit pb) qui fait référence à cette complémentarité.
Par exemple : ATCG est complémentaire de TAGC.
ATCG
||||
TAGC
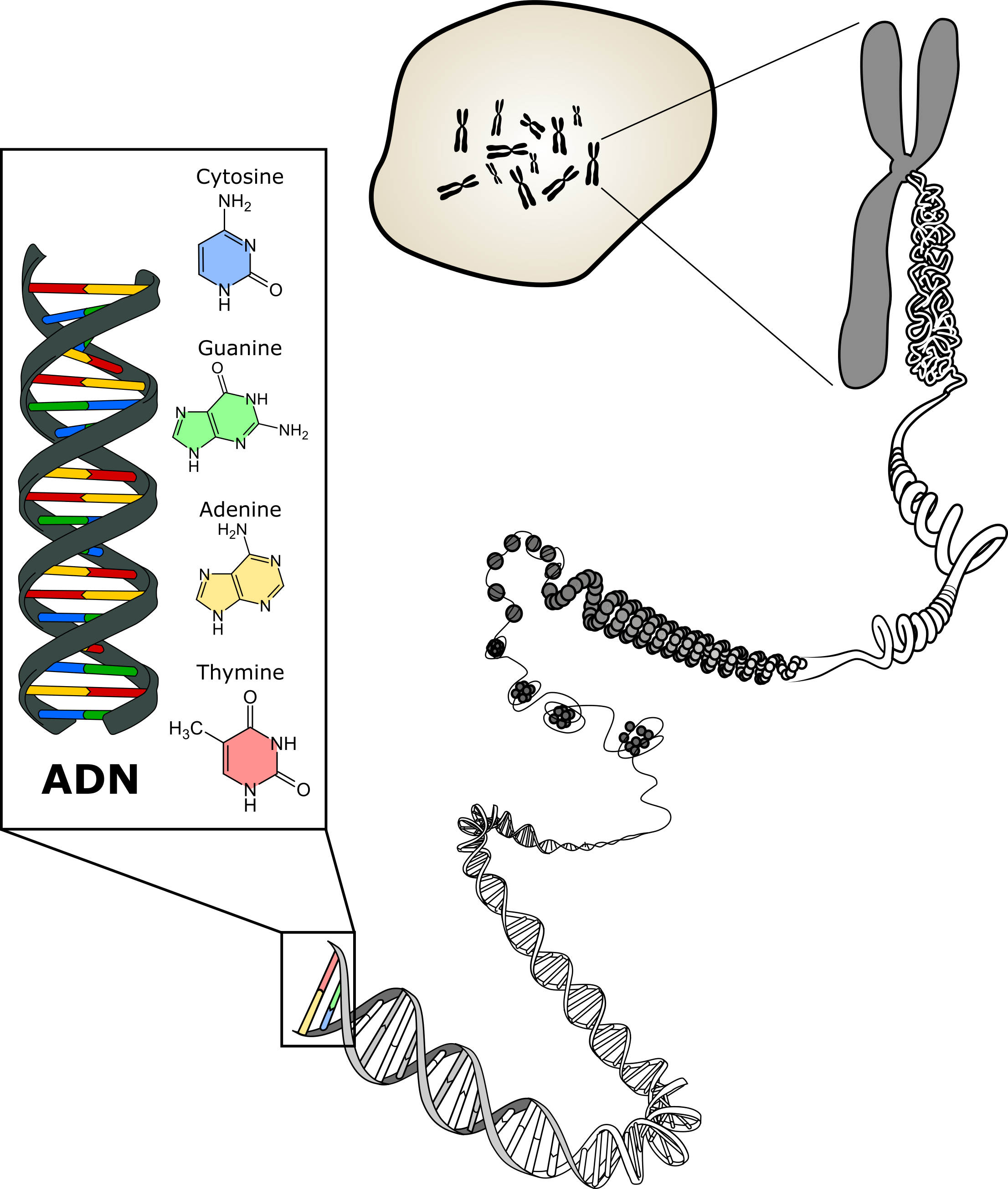
Tous les organismes cellulaires (bactéries, animaux, plantes, champignons, etc) ont de l’ADN mais le génome est différent pour chaque espèce, nous n’avons pas le même plan que la banane. Cela dit, certaines parties du plan sont communes à presque tous les organismes. Il y a des fonctions que toutes les cellules doivent pouvoir effectuer pour survivre, par exemple savoir lire le génome. Autrement dit, les parties du plan qui servent à produire les outils pour lire le plan sont presque universelles dans le monde vivant.
Vous connaissez probablement un autre nom pour les "parties du plan", il s'agit des gènes. Il n'y a pas de définition absolument consensuelle de ce qu'est un gène mais la plus classique est qu'il s'agit d'une portion d'ADN qui a un rôle dans la production d'une protéine. Les protéines sont les outils de la cellule, ce sont elles qui permettent toutes les fonctions des cellules et je ne peux pas rentrer dans le détail de toutes les fonctions des protéines car il y a littéralement écrit des livres de centaines de pages dédiés uniquement au sujet des protéines. Tout ce qu'il faut savoir c'est que le plan de fabrication des protéines est appelé gène et qu'il se trouve sous forme de molécule d'ADN dans le génome.
Le génome de Adineta vaga (parce que oui c'était le titre quand même)...
Le travail présenté dans l'article scientifique publié a été de reconstruire le génome de A. vaga. Notre but est de savoir s’il peut nous aider à comprendre pourquoi A. vaga est asexué ? Mais aussi savoir s'il y a des indications dans ce génome qui expliquent pourquoi A. vaga survit à des conditions extrêmes ?
D'anciens travaux déjà publiés ont présentés des pistes de réponses à ces questions. Par exemple, on a pu démontrer qu’en avoir irradiant A. vaga son génome est complètement fragmenté mais les cellules de cet animal sont capables après quelques heures ou quelques jours de recoller les morceaux. Une cellule humaine soumise à un traitement similaire serait simplement morte.
Un autre article publié en 2013 a été extrêmement important pour la communauté scientifique qui étudie les rotifères et l'asexualité en général. Cet article présente une explication de pourquoi Adineta vaga est asexué.
La raison que l'article présente est simple mais elle nécessite d'avoir quelques bases en reproduction. La plupart des animaux sexués ont deux copies de leur génome, une copie qui provient de la mère et une copie qui provient du père. Normalement, ces copies se rencontrent lors de la fécondation d’un ovule et d’un spermatozoïde qui apportent chacun une moitié de génome, le spermatozoïde contient la moitié du génome du père et l’ovule la moitié de celui de la mère. Lors de la fécondation, les copies fusionnent et on obtient un embryon qui commence à se développer.
Par conséquent, une étape cruciale est de produire les spermatozoïdes et les ovules. Ce sont des cellules particulières car elles ne contiennent que la moitié du génome alors que toutes les autres cellules contiennent le génome entier.
Il y a deux mécanismes qui permettent aux cellules de se diviser. Les mitoses et les méioses. La vaste majorité des divisions cellulaires sont des mitoses.
Les mitoses sont des divisions qui se font en trois étapes :
- Copier le génome.
- Compacter le génome en chromosomes.
- Se diviser en deux parties chacune avec une moitié du génome.
Avant l’étape 2, les chromosomes sont décondensés et le génome ressemble à un plat de spaghettis sauf que pour un humain il n’y aurait que 46 longs spaghettis. La compaction en chromosome permet de démêler le plat de spaghettis. Elle est importante pour que les cellules qui s’engagent dans une division s’assurent de transmettre tout le plan à chacune des deux cellules filles qui résultent de la mitose.
La vidéo suivante montre des mitoses dans un embryon d’oursin. Ici l’ADN a été coloré artificiellement en jaune et les microtubules en bleu. Les microtubules sont une sorte d’échafaudage interne à la cellule qui permettent notamment de séparer les chromosomes (l’ADN condensé). Vous pouvez voir la différence avant et après la compaction de l’ADN dans les cellules en haut à droite de la vidéo, l’ADN est d’abord diffus (plat de spaghettis) puis devient compact (forme de bâtons). En bas à gauche l’ADN est déjà condensé et la cellule est prête à se diviser.
Les méioses sont rares car elles ne servent qu’à produire les ovules et les spermatozoïdes. Ces cellules ne contiennent que la moitié du génome. Comme vous le savez maintenant, on reçoit deux copies du plan, une copie paternelle via le spermatozoïde et une copie maternelle via l’ovule. On dit que les cellules avec ces deux copies sont « diploïdes » et on note ces cellules 2n2c. 2n car elles contiennent 2 versions du plan et 2c car il y a une copie de chaque version (2x1c). Lors de l’étape 1 d’une mitose, les deux versions sont copiées une fois et on a donc 2 copies de chaque version, on dit que la cellule est 2n4c (2 versions et 2 copies par version donc 2x2c = 4c au total).
La plupart des cellules sont 2n2c. Alors que les ovules et les spermatozoïdes sont 1n1c : une seule version et une seule copie par version.
On peut donc décrire décrire une méiose comme ceci :
- Une cellule de départ : 2n2c
- Copie de chaque version : 2n4c
- Première division (méiose I) : on obtient 2 cellules 1n2c
- Deuxième division (méiose II) : on obtient 4 cellules 1n1c
Si vous avez bien suivi vous remarquerez qu’on pourrait s’en sortir plus facilement en ne copiant pas l’ADN au départ. Si on faisait directement étape 1 -> 3, on passerait de 2n2c à deux cellules 1n1c.
Evidemment, la sélection naturelle a conservé ces étapes supplémentaires et cette façon plus compliquée de produire les ovules et les spermatozoïdes pour une bonne raison. En effet, ça a un coût énergétique de produire quatre cellules au lieu de deux. Il doit y avoir un avantage évolutif à quand même passer par ce surcoût énergétique.
En fait, si on fait une copie, on peut ensuite mélanger les versions parentales. Il y a apparemment de gros avantages à faire ce mélange des deux versions parentales car presque tous les animaux le font. Je ne rentrerai pas dans tous les détails des avantages mais il s'agit principalement de créer de la diversité génétique. Tous les membres d'une famille ne se ressemblent pas trait pour trait et c'est en bonne partie un résultat de la recombinaison.
Vous pouvez voir une méiose sur cette image (je n’ai pas trouvé de vidéo claire avec une bonne qualité). Il faut lire par colonne d'abord la première puis la seconde. La couleur rouge indique la position des chromosomes.
. https://doi.org/10.1186/s12870-018-1442-y")
On peut d’abord voir la condensation du génome en chromosomes (a2 - b2). Ensuite les chromosomes se rassemblent (b2 - c2). Á l'étape suivante (c2 - d2), les chromosomes paternels se sont collés à leurs homologues maternels. On a donc l’impression sur les photos c2 et d2 qu’il n’y que la moitié des chromosomes par rapport à l'étape b2 en réalité la moitié n'a pas disparu mais ils se sont appariés.
Pendant cet appariement, les chromosomes homologues recombinent, c’est à ce moment qu’il y a mélange entre les versions paternelle et maternelle du génome. Les chromosomes homologues s'échangent littéralement des morceaux d'ADN, physiquement la molécule est coupée puis recollée sur une autre. Le schéma suivant illustre les trois premières étape de la première division de la méiose (sur l'image ci-dessus, ce sont les étapes b2 à e2).
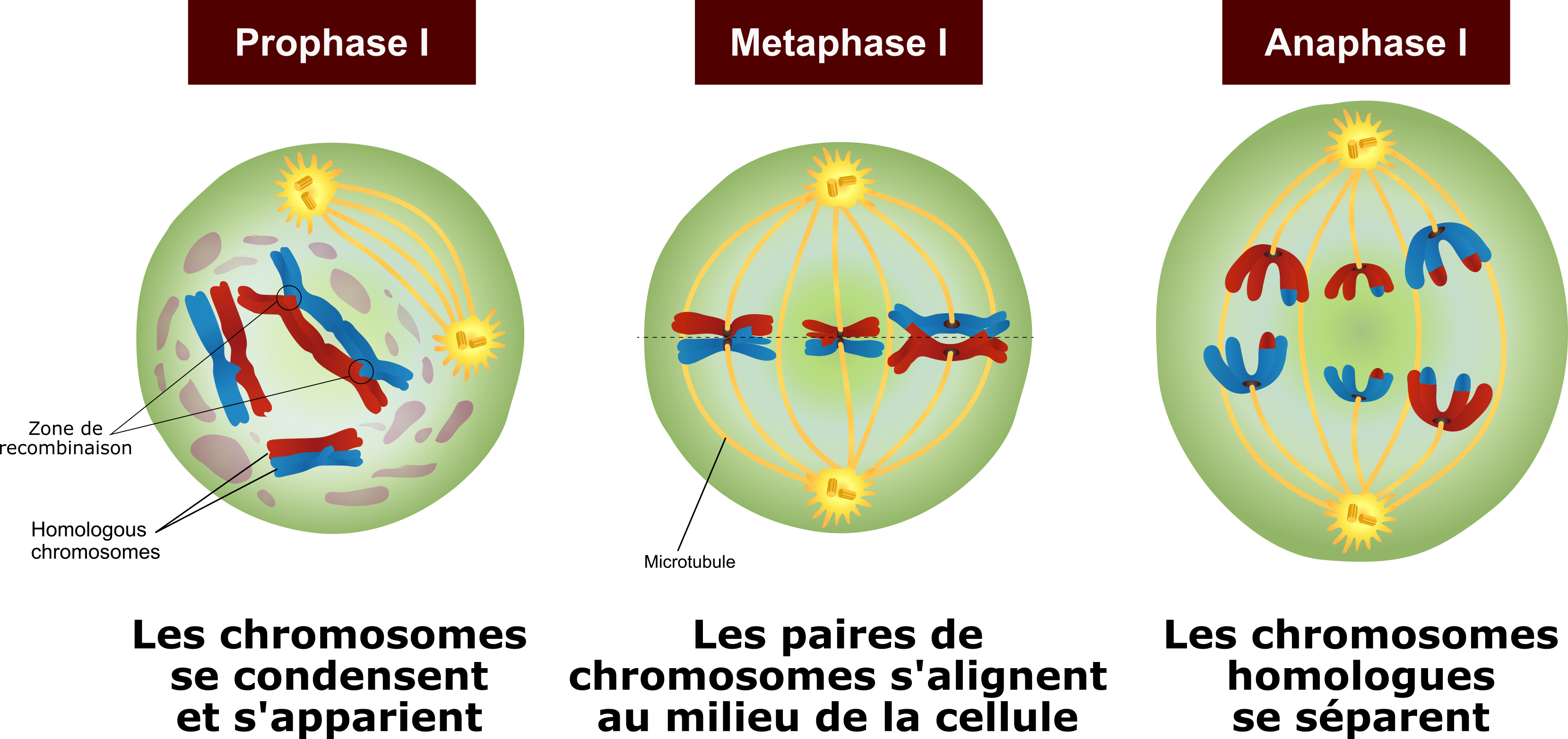
Après la première division qui sépare les paires de chromosomes homologues, les cellules sont désormais 1n2c. A ce stade, on a une seule version du génome et chaque chromosome est composé de 2 copies qu'on appelle les chromatides soeurs. La méiose continue avec une deuxième division (visible sur la figure des microscopies ci-dessus, il s'agit des étapes f2 à j2), lors de cette étape, ce sont les chromatides soeurs qui se séparent. Les cellules sont désormais 1n1c, elles vont continuer à se développer en ovules ou spermatozoïdes.
L'origine de la confusion
L'article publié en 2013 propose une explication de pourquoi A. vaga est asexué. L'article démontre que le génome de A. vaga ne contient pas de chromosomes homologues. En bref, en observant leurs résultats, on trouve bien 2 copies de chaque gènes comme s'il y avait deux versions (paternelle et maternelle chez les organismes sexués) mais il n'y a pas deux chromosomes similaires, les chromosomes n'ont pas la même structure et donc ils ne peuvent pas s'apparier ni recombiner.
C'est une découverte importante car la seule du genre et à en 2013, elle semble bien expliquer la raison de l'asexualité, cette découverte explique qu'on ne peut pas faire de méiose car il n'y a pas de paires de chromosomes homologues. Pas de chromosomes homologues, pas d'appariement, pas de recombinaison. En bref, pas de méiose possible. Le schéma ci-dessous montré un exemple de la structure supposée du génome de Adineta vaga. Les couleurs indiquent les zones homologues capables de s'apparier sur les chromosomes.

Ce n'est malheureusement pas aussi simple!
Suite à la publication de ce résultat, l'hypothèse est que A. vaga se reproduit en utilisant exclusivement des mitoses. Cependant, il reste des inconnues. Un point étrange est l'impossibilité de faire de la recombinaison. En temps normal, la recombinaison permet aussi de réparer les cassures de l'ADN en utilisant le chromosome homologue qui n'est pas endommagé. Cependant, s'il n'y a pas de chromosomes homologues, ça devient difficile de faire de la recombinaison et de réparer ces cassures. Pourtant Adineta vaga est très résistant aux cassures de l'ADN.
Un autre point étrange est que si A. vaga ne se reproduit qu'avec des mitoses, on s'attend qu'avec le temps on observe des mutations qui s'accumulent indépendamment entre les copies de gènes. Pourtant on retrouve des copies bien conservées de gènes voire des copies identiques.
Suite à la publication du génome de 2013, plusieurs hypothèses ont été avancées pour répondre à ces questions. Cependant, aucune n'est prouvée à cette époque. Il faut donc tester tout ça.
Séquençage et assemblage de génomes
Depuis 2013, les technologies ont évolué, d’autres études sont arrivées et le doute a commencé à s’installer sur la structure étrange du génome.
Nous avons donc reséquencé le génome de A. vaga. En pratique, on a extrait l’ADN des cellules. Ensuite on a envoyé cet ADN extrait à des labos spécialisés dans le séquençage (la lecture de l’ADN).
Il y a plusieurs technologies qui permettent de lire l'ADN. En général il y a deux aspects dans ces technologies : la longueur des séquences lues et la précision des séquences lues.
Nous avons reçu des séquences de nucléotides très précises (qui contiennent très peu d’erreurs) mais ces séquences sont relativement courtes (environ 250 paires de bases lues).
Pour vous donner une idée, le génome de Adineta vaga est assez petit, il contient environ 200Mb (diploïde donc 2n). On peut s’en tirer plus facilement en cherchant à reconstruire un « consensus » de ces deux versions du génome. Peu importe si on se trompe parfois en fusionnant une partie du génome maternel avec une partie du génome paternel. Comme ces assemblages se ressemblent on peut les fusionner. Dans le cas de Adineta vaga, c’est environ 1,7% de différence entre ces deux versions. On se facilite un peu la tâche en reconstruisant un génome 1n au lieu de 2n et donc assembler 100Mb.
La tâche reste difficile avec des séquences courtes (250pb) pour reprendre l'analogie des bibles, c'est comme si on avait pris un livre d'une taille de 27 bibles et que nous avions reçu des phrases de 10 mots pour reconstruire ce livre de 27 équivalents-bibles sans se tromper dans l'ordre des mots.
Il y a des algorithmes très puissants qui permettent de reconstruire le génome à partir de ces séquences et d’obtenir de longs fragments de génome assemblés. Malheureusement, les séquences courtes ne suffisent pas à obtenir un assemblage avec une structure complète. En appliquant les algorithmes, on arrive à obtenir des pages ou des chapitres entiers mais on ne reconstruit pas le livre entier, il reste beaucoup de trous et on ne connait pas bien l’ordre ni l’orientation des pages (ici l’analogie avec un livre ne fonctionne pas car on lit de gauche à droite et de bas en haut mais cette propriété ne s’applique pas à l’ADN, les séquences sont symétriques et on ne peut pas savoir dans quelle ordre et quelle orientation des fragments indépendamment sont les uns par rapport aux autres).
En fait, il y a des obstacles pratiquement insurmontables qui empêchent l’assemblage avec des petites séquences. Par exemple, s’il y a des séquences répétées plus longues que 250 nucléotides, si la même phrase de plus de 10 mots se retrouve plusieurs fois dans le livre à des endroits différents, on sera obligé de couper l’assemblage au niveau de ces régions car aucune séquence n'est suffisamment longue pour connaitre à la fois ce qu'il y a avant et après la région répétée. Ce problème est illustré dans la figure ci-dessous.
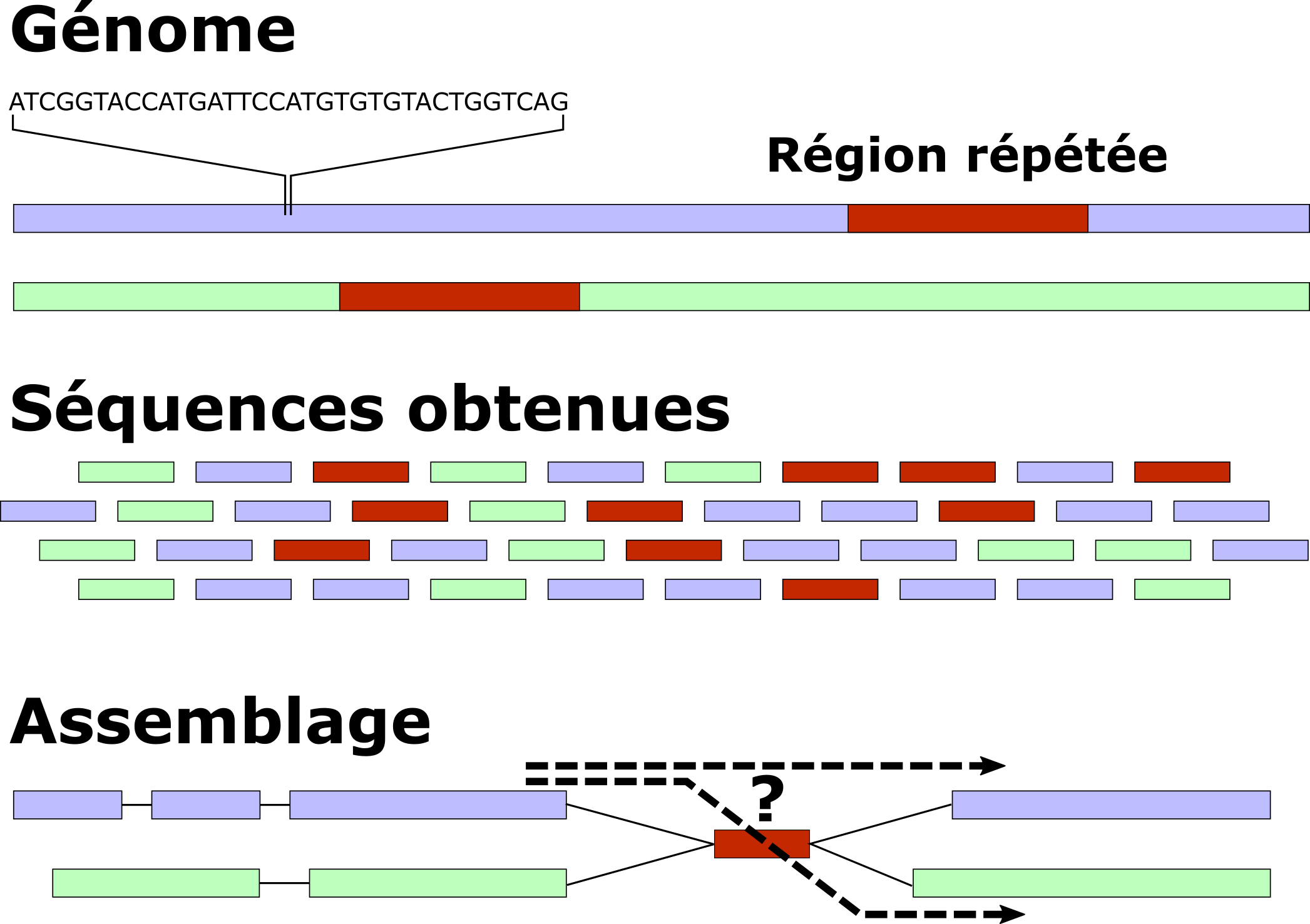
Évidemment, des ingénieurs et des scientifiques ont développé des technologies afin de surmonter ces obstacles. Nous avons donc pu obtenir deux autres types de séquences : des séquences longues (grâce aux technologies développée par Pacific Bioscience © et Oxford Nanopore ©) et des séquences dont on connaît la proximité physique.
Les séquences longues sont comme des demi-pages complètes mais qui contiennent beaucoup d’erreurs, elles peuvent couvrir entre 1000 et 1000,000 nucléotides mais environ 10 à 15% des nucléotides sont faux ou peu fiables. Avec ces séquences, on arrive à obtenir de longs fragments bien assemblés mais qui sont rempli d’erreurs au niveau de la séquence spécifique. Malgré tout, il reste des régions compliquées où les algorithmes d'assemblages ont besoin d’informations avec encore une échelle de distance supplémentaire pour obtenir un assemblage complètement résolu.
Pour obtenir cette information, on peut utiliser encore une autre technologie de séquençage d’ADN. On souhaite connaître la proximité physique entre deux séquences. Pour faire ça, on peut séquencer l’ADN par « paire » grâce à une technologie qui s’appelle HiC (prononcé aïe-scie).
L’idée du HiC est de figer l’ADN dans les cellules grâce à des composés chimiques on peut « coller » l’ADN. Dans une cellule, l'ADN peut être plus ou moins condensé. Cette condensation est permise par des protéines qui maintiennent l’ADN dans une certaine conformation physique. Lorsqu’on colle l’ADN, on crée des liens entre l’ADN et ces protéines de structure. Ensuite, on va « digérer » l’ADN qui n'est pas collé à des protéines grâce à des enzymes (un autre type de protéines). Certaines enzymes se spécialisent dans la digestion des brins d’ADN. Une fois que l’ADN est digéré, il reste les protéines attachées à des fragments d’ADN. On va maintenant utiliser encore un autre type d'enzymes qui attachent les extrémités libres d’ADN. Ces enzymes (les ligases) vont lier les fragments d'ADN proches physiquement. On obtient des fragments d’ADN circulaire. On va les décoller des protéines puis les récupérer. Il suffit maintenant de lire les séquences et on peut avoir une idée de quelles séquences sont physiquement proches les unes des autres. Comme statistiquement les séquences d’un même chromosome sont plus souvent proches entre-elles que les séquences de deux chromosomes différents, la majorité des ADNs circulaires obtenus correspondent à des régions d’ADN d’un même chromosome. Le schéma suivant illustre le processus pour obtenir des données HiC. Notez qu'en plus de permettre de dire quels fragments de l'assemblage vont ensemble, les données HiC permettent de connaitre l'orientation relative des fragments les uns par rapport aux autres.
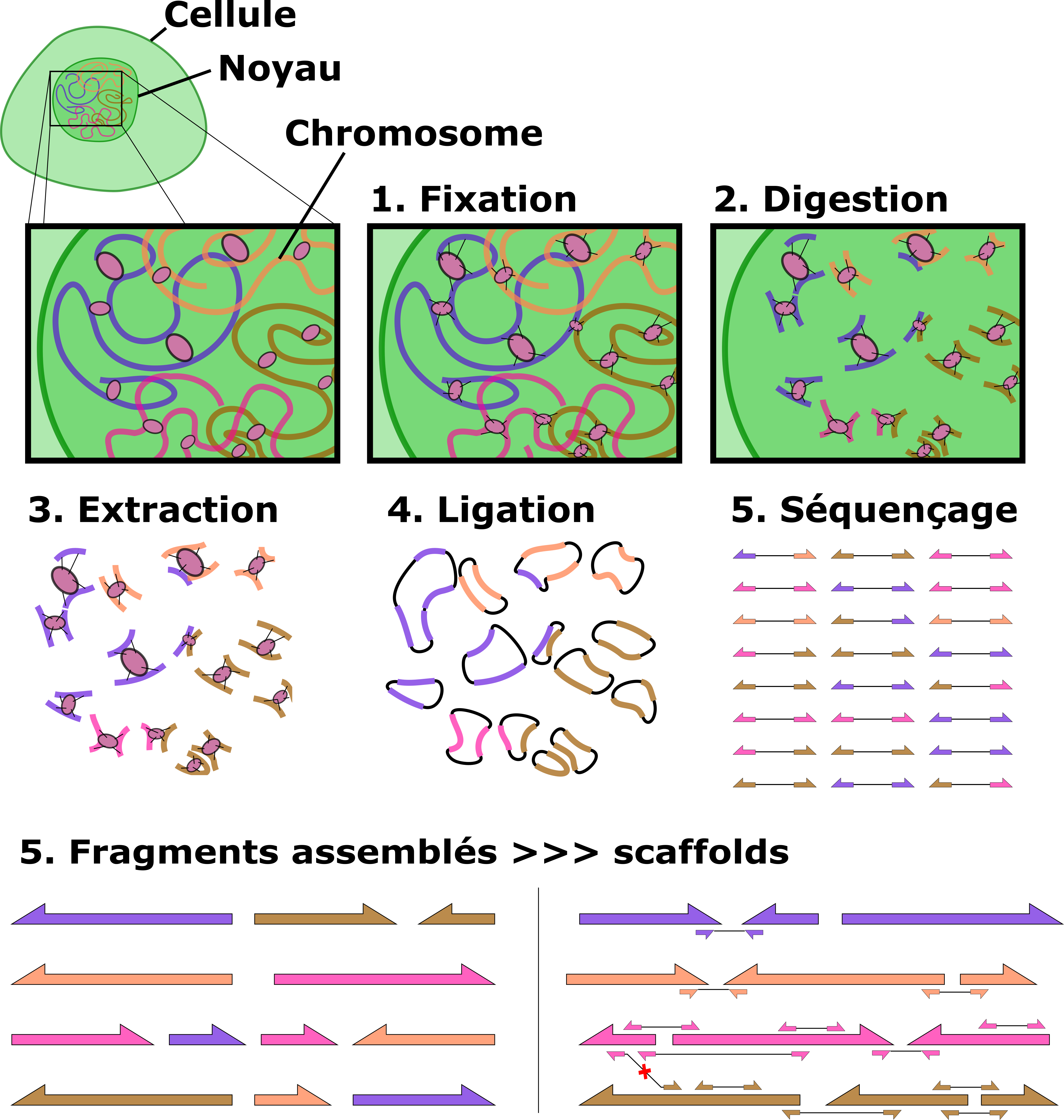
Recette pour 1 assemblage de génome de Adineta vaga
On a 3 types de séquences, des séquences courtes et précises, des séquences longues et imprécises et des séquences pairées dont on connait la proximité physique. Maintenant, il reste à combiner tout ça pour reconstruire le génome...
J’ai beaucoup exploré différentes méthodes d'assemblages pour trouver un processus idéal avec ces données. D'ailleurs avec l’aide de plusieurs collègues et de ces données nous en avons profité pour tester extensivement les différents programmes d'assemblages.
Finalement nous avons gardé une méthode satisfaisante. Ce n'était pas la seule possibilité et d'autres approches étaient prometteuses mais après beaucoup de comparaisons, discussions et différents tests nous sommes tombés d'accord sur la suivante :
- Utiliser les longues séquences pour obtenir la structure des chromosomes. Ces séquences sont encore un peu imprécises au niveau des nucléotides individuels.
- Utiliser un outil qui détecte les erreurs dans ce premier assemblage. Une des erreurs fréquentes dans notre cas était que l’assemblage de départ contient parfois deux versions de certaines régions (2n) et parfois une seule version (1n). Comme nous essayons de reconstruire un génome 1n, il fallait donc détecter quelles régions étaient présentes en deux versions pour n'en garder qu'une seule.
- Utiliser un outil qui corrige les erreurs au niveau des nucléotides. Les séquences courtes sont plus précises et contiennent peu d’erreurs, on peut les aligner sur la structure du génome assemblé et corriger les erreurs au niveau des nucléotides en comparant l'assemblage aux séquences courtes alignées.
- Finalement, il nous reste quelques fragments (environ une vingtaine à ce stade) et on ne sait pas comment attacher et orienter ces fragments entre eux. Nous avons donc utilisé les données HiC pour savoir comment coller et orienter les fragments entre eux.
Cet assemblage n’est que la première étape. Je me suis principalement chargé de faire l’assemblage que je décris ci-dessus mais les collègues ont utilisé d’autres approches pour confirmer cette structure. Une première confirmation vient d'autres outils d'assemblages. Un premier outil essaie d’assembler un génome 2n (il essaie d’assembler les versions paternelles et maternelles séparément) à partir uniquement des longues séquences et des données HiC. Un second outil essaie juste d’assembler les séquences courtes uniquement, ensuite on a utilisé les données HiC pour obtenir la structure.
Quand on compare ces autres assemblages à celui que j'ai décrit plus en détail, ils donnent tous le même résultat concernant la structure du génome, ils sont aussi très largement d’accord sur les séquences précises : c’est donc une bonne confirmation du premier assemblage.
Retour à la biochimie!
Toutes ces analyses sont "bioinformatiques" ce qui signifie qu'elles sont principalement réalisées dans l'ordinateur. Même si j’ai personnellement confiance dans tous ces résultats, mon but n’est pas de me convaincre moi-même mais surtout de convaincre les autres scientifiques du domaine que notre résultat est correct et donc que notre travail a une valeur pour toute la communauté.
On veut une preuve sans appel et pour ça, il faut utiliser des méthodes qui ne dépendent pas du séquençage et d’assemblages mais qui indiqueront tout de même si l'assemblage qu'on propose est correct… Maintenant qu'on pense avoir la bonne structure et la bonne séquence des différents chromosomes on peut aller voir directement dans la cellule si les molécules d’ADN qui correspondent à ces séquences sont d'accord avec nos assemblages.
Pour faire ça, on va utiliser une méthode appelée « FISH » (qui signifie Fluorescence In Situ Hybridization). Comme l’ADN est composée de 2 brins complémentaires, on peut exploiter cette caractéristique pour connaître l’emplacement de certaines séquences sur les chromosomes. En effet, si on introduit un brin d‘ADN avec une séquence complémentaire et qu’on chauffe l’échantillon (pour séparer les deux brins originaux), on peut faire rentrer le brin d’ADN qu’on a introduit dans la molécule d’ADN qui compose le chromosome. On appelle ce processus « hybridation » parce que deux ADNs simples brins s’hybrident pour former un ADN normal (double brin).
On va donc synthétiser (en vérité, on délègue ce travail à des firmes spécialisées) des séquences d’ADN plus ou moins longues (entre 20 et 100 nucléotides) qui correspondent à différentes régions de notre assemblage. On appelle ces séquences des sondes. Nous avons choisi 6 régions (environ, 2 millions de paires de bases - Mbps), sur 3 paires de chromosomes. Pour chaque paire de chromosome, on a choisi 2 régions : une à chaque extrémité. Dans chacune de ces régions, on va synthétiser quelques centaines de sondes. On obtient donc 6 « librairies » (3 paires de chromosomes x 2 régions par paire) avec quelques centaines de sondes étalées sur 2 Mbps.
On va ensuite attacher des molécules fluorescentes sur ces sondes et séparer les deux brins d’ADN de chaque sonde. Ensuite, on va insérer ces sondes dans une cellule de A. vaga. Il suffit ensuite de chauffer les cellules afin de séparer les doubles brins des chromosomes et permettre à nos sondes de s’hybrider aux niveaux de leurs séquences complémentaires. Comme les sondes sont fluorescentes, on va utiliser un microscope spécial pour détecter l'emplacement des molécules fluorescentes.
On veut aussi savoir où sont les chromosomes, on va donc ajouter des molécules fluorescentes (d’une autre couleur) qui vont aller colorer tout l’ADN de la cellule sans distinction de la séquence choisie. On a utilisé une molécule appelée DAPI pour faire ça.
La plupart des cellules ont de l’ADN décondensé, pour être sûr de la structure, on veut que les librairies de sondes fluorescentes soient bien distinctes et placée sur des chromosomes bien séparés des autres. Une façon de faire ça, c’est d’aller chercher des cellules où les chromosomes sont condensés. Il y en a relativement peu chez les individus adultes, par contre, dans les œufs récemment pondus, on sait que les chromosomes sont condensés car ils sont composés d’une seule cellule. Comme cette cellule va obligatoirement commencer à se diviser, on peut être quasiment sûr que les chromosomes sont condensés dedans.
Qu'est ce qu'on s'attend à voir ? Si nos séquences sont mal assemblées soit on ne verra aucune fluorescence parce que les sondes ne se sont pas hybridées, ce qui signifie que la séquence des sondes est fausse et donc notre assemblage l'est aussi. Soit on verra de la fluorescence à plusieurs endroits sur plusieurs chromosomes, ce qui signifie que la structure de notre assemblage est fausse car les sondes d'une même librairie ne se sont pas toutes mises au même endroit proches les unes des autres.
Si nos séquences sont bien assemblées, ce qu’on espère voir, c’est que toutes les sondes d’une librairie s’hybrident sur les deux chromosomes homologues. Comme on a fait deux librairies une à chaque extrémité des chromosomes d’une même paire on devrait voir 2 chromosomes avec 2 points de couleurs différentes (ici rouge et vert) et les couleurs ne doivent pas être superposées car les librairies sont aux extrémités.
Comme vous le constatez sur la photo de microscopie, on obtient le résultat attendu. Il confirme que nos assemblages sont corrects.



Le nouveau génome de Adineta vaga
La figure ci-dessous montre une représentation schématique du génome avec le nombre réel de chromosome dans les cellules (2n2c). En réalité l’assemblage principal ne contient que 6 séquences et non 12 car les paires de chromosomes homologues sont représentés par une seule séquence consensus. Le fichier texte contenant l’assemblage est donc 1n et pas 2n.
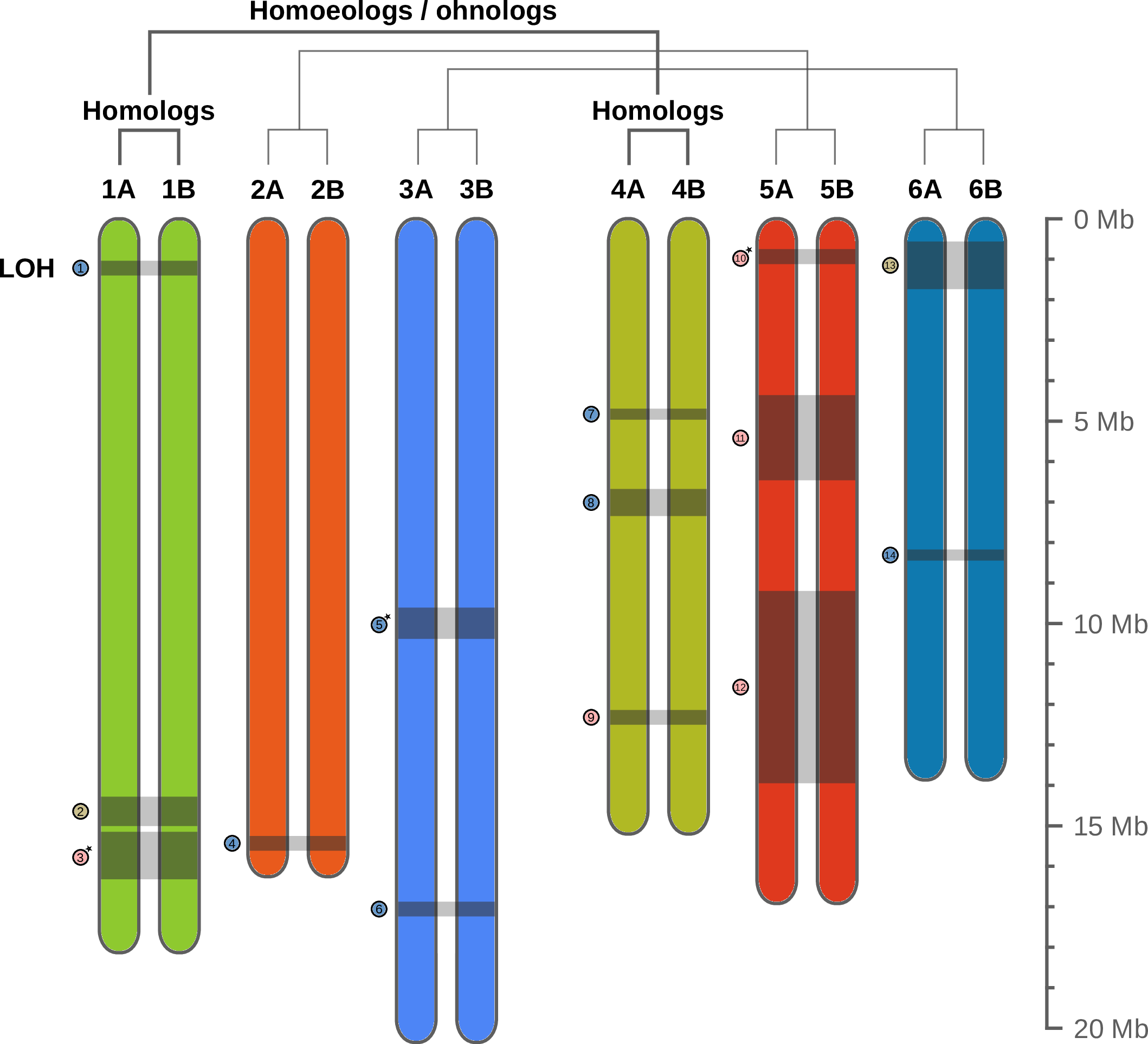
Vous noterez qu’il y a deux types de liens entre les chromosomes. Il y a des paires nommées « homologs » qui correspondent aux paires de chromosomes homologues (le génome est 2n) et les chromosomes sont notés A ou B au sein d'une même paire.
Il y a aussi des groupes de 4 chromosomes nommés « homoeologs / ohnologs », ces mots indiquent que ces chromosomes sont apparentés. On pense que dans l’histoire évolutive de la lignée de A. vaga, un ancêtre distant ne possédait que 3 paires de chromosomes par exemple 1,2,3 ou 4,5,6 mais en réalité n’importe quelle combinaison entre [1 / 4], [2 / 5] et [3 / 6] est possible.
On sait que ces chromosomes sont apparentés car on peut détecter que leurs séquences se ressemblent. On sait déjà que la différence entre les séquences homologues (entre 1A et 1B par exemple) est relativement faible (environ 1,7% des bases sont différentes). Comme la différence est faible on peut créer une séquence consensuelle qui représente autant le chromosome A que son homologue B.
Dans l'article on appelle la propriété d'avoir des séquences homologues proches (1.7% diff) et des séquences homoeologues éloignées (~15% diff) paléotétraploidie. "Paléo" parce qu'on pense qu'il s'agit d'un événement ancien et "tétraploidie" parce qu'il y a 4 versions différentes qui ont évolué à partir d'une séquence ancestrale.
Pour connaître la différence entre les chromosomes homologues, il suffit de prendre la séquence du chromosome assemblé et les séquences courtes et précises qui ont servi à obtenir l’assemblage. On a aligné ces séquences sur l’assemblage. Ensuite, on a scanné le long du chromosome et on a répertorié tous les endroits où dans les séquences courtes alignées on retrouve deux versions différentes qui correspondent aux différences entre les homologues. Il suffit donc de compter le nombre total de différences puis de diviser par la taille du génome (1n). Dans notre cas, le génome fait à peu près 100 Mbps (100 millions de paires de bases) et on retrouve à peu près 1,7 millions de sites où il y a deux versions différentes dans les séquences brutes. Ca nous donne donc 1,7 millions / 100 millions = 1,7% de bases sont divergentes entre les séquences des chromosomes homologues. La figure ci-dessous illustre ce processus.

Pour détecter la divergence entre les chromosomes homoeologues, on peut utiliser une approche similaire mais c’est un poil plus difficile car les séquences brutes ne vont pas s’aligner à deux endroits différents. Par exemple, une séquence provenant du chromosome 1A va s'aligner sur le consensus du chromosome 1 mais pas sur la séquence consensuelle du chromosome 4 même si les séquences se ressemblent un peu. Comme les séquences brutes proviennent soit d’une version (A ou B) du chromosome 1 ou d’une version du chromosome 4, elles seront toujours plus proches de 1 ou de 4 et vont s’aligner de préférence sur la séquence la plus proche mais pas sur les deux en même temps.
Donc on ne peut pas compter aussi simplement ces différences. Par contre, on peut essayer d’aligner les séquences des chromosomes sur les autres chromosomes. Certains algorithmes sont spécialisés dans ce genre d’alignements et on trouve que la séquence consensus du chromosome 1 s’aligne sur celle du chromosome 4, le chromosome 2 s'aligne avec le 5 et le 3 avec le 6. En utilisant cette approche, on trouve que les homoeologues ont environ 10 à 20% de divergence. Ce qui signifie que tout de même 80% de l’entièreté des séquences sont identiques. Malheureusement ce nombre n’indique pas si le séquences sont dans le même ordre et en réalité il y a des réarrangements dans l’ordre des séquences, des inversions, des pertes et des gains de nouvelles séquences. Pour reprendre l’analogie d’un livre, les phrases sont 80% identiques mais l’ordre des paragraphes et de certains chapitres est différent.
Pour vérifier si la structure des chromosomes homoeologues est la conservée, on doit aller chercher au niveau des gènes. Mais pour bien comprendre pourquoi dans les gènes, on va devoir parler de mutations et de sélection.
Comme les gènes sont impliqués dans la production des protéines et que les protéines sont absolument nécessaire au bon fonctionnement de la cellule. Les gènes ne peuvent pas muter n'importe comment. Des mutations apparaissent spontanément sur les chromosomes quand l'ADN est copié avant une division cellulaire. Mais toutes les mutations ne sont pas équivalentes. La plupart des mutations ne font rien et la séquence d'ADN change sans que ça impacte l'organisme de quelque façon que ce soit. Ce sont des mutations neutres.
Parfois les mutations sont bénéfiques. Par exemple, une mutation qui permet à une plante de grandir quelques centimètres de plus que ses voisines lui permettra par la même occasion de capter plus de soleil et donc de mieux se nourrir et survivre que les plantes voisines. Elle aura donc plus de chance que les autres de se reproduire et la nouvelle mutation aura plus d'opportunité de se propager parmi ses descendants. On appelle ce processus différence de succès reproductif mais vous l'avez probablement entendu sous un autre nom : la sélection naturelle. On l'appelle comme ça parce qu'en pratique c'est comme si la nature sélectionnait les individus qui se reproduisent le mieux dans leur environnement. En réalité, il n'y a personne qui sélectionne, c'est la différence de succès reproducteur de la plante par rapport à ses voisines qui fait que la mutation va se propager dans la population. En bref, les mutations arrivent au hasard mais les individus qui ont la chance d'avoir une mutation bénéfique vont mieux se reproduire (être sélectionné) et se retrouver plus fréquemment que les autres à la génération suivante.
Le corolaire est vrai également, si la nouvelle mutation est délétère pour l'organisme, si par exemple elle l'empêche la plante de grandir, alors la plante ne captera plus autant de soleil que ses voisines, elle va donc moins être capable de se reproduire et la mutation va peu à peu disparaître de la population car il y aura peu de descendants de la petite plante et beaucoup de descendant de la grande plante.
Les mutations bénéfiques et délétères ont généralement lieu dans les gènes car ils contribuent à la production des protéines et certaines protéines sont essentielles au bon fonctionnement des organismes. Une mutation qui empêche un gène de produire une protéine fonctionnelle va être délétère et va donc avoir moins de chance de se retrouver dans une population. Certaines mutations sont carrément léthales et les individus qui les portent meurent avant de se reproduire, par exemple, l'embryon qui porte cette mutation ne se développe même pas.
En bref, les gènes ne peuvent pas accumuler autant du mutation que les séquences en dehors des gènes (les séquences qui n'interviennent pas dans la production de protéines). Avec Adineta vaga, on peut facilement observer l'effet de la sélection sur les mutations. En comparant les mutations dans les gènes on trouve que la différence moyenne entre les homologues est de ~0.5% (5 mutations pour 95 bases identiques) alors que quand on regarde en dehors des gènes on trouve que la différence moyenne est de ~1.7% (17 mutations pour 83 bases identiques). C'est parce que les gènes ne peuvent pas muter aussi facilement, on risque toujours de "casser" les gènes et donc de dérégler la production d'une protéine.
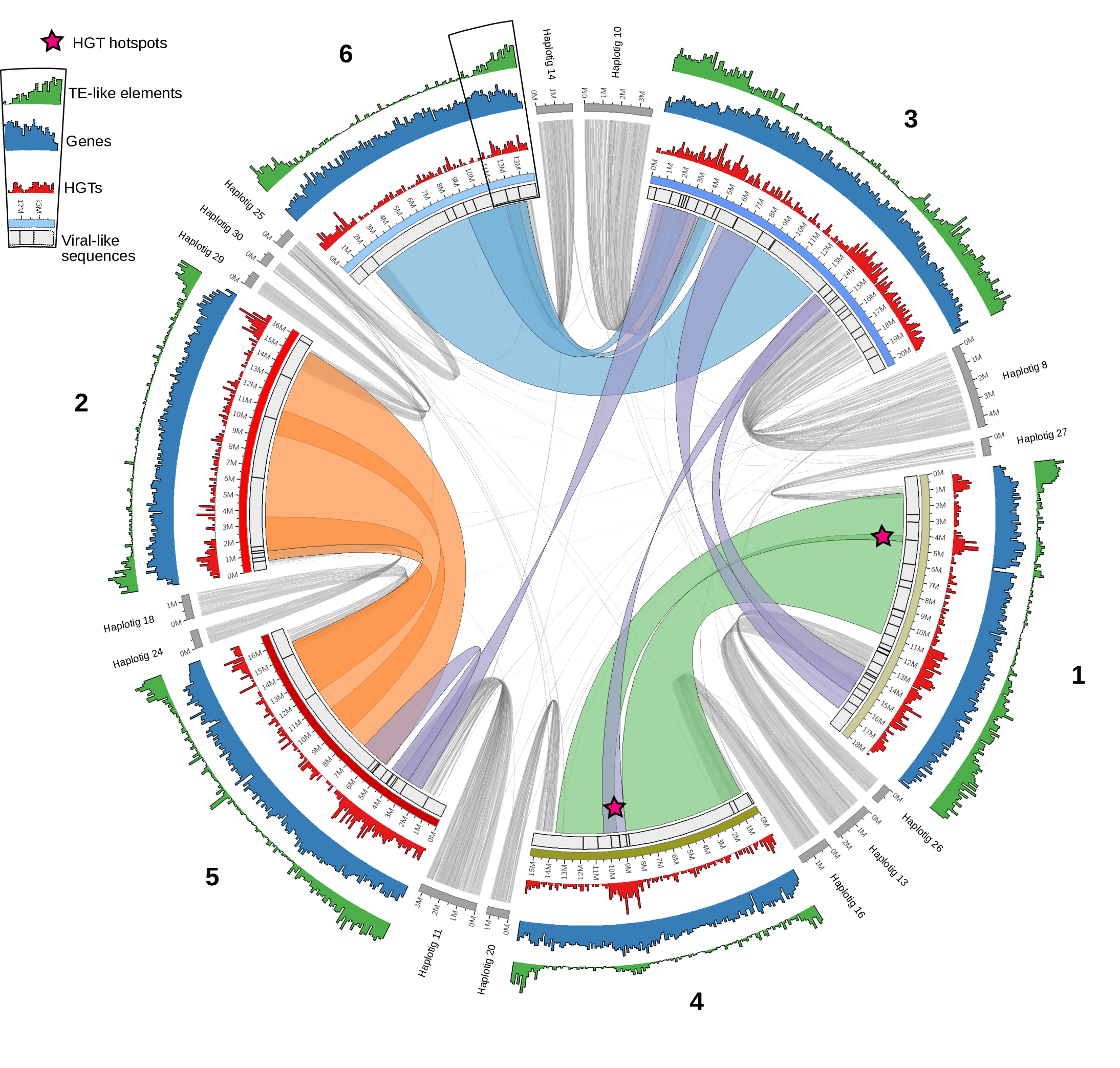
Si vous avez bien suivi, c'est la raison pour laquelle on va regarder dans les gènes, on s'attend à ce que la divergence entre les gènes homoeologue soit plus faible que la divergence moyenne entre les séquences intergéniques (entre les gènes) homoeologues. On a donc détecter tous les gènes qui se ressemblent sur les homoeologues et on a essayé d'observer si leur ordre est le même, si la structure des chromosomes est conservée entre les homoeologues. La figure suivante vous montre ce résultat. Les chromosomes sont représentés par des barres circulaires, il y en a 6 en tout, une par paire de chromosomes homologues.
La figure est un peu compliquée donc pour simplifier, vous pouvez ignorer les graphiques rouge bleu et vert sur les cercles extérieurs ainsi que les liens gris clair. Ce qui nous intéresse ce sont les liens au milieu de la figure en bleu clair, orange et vert clair. Ces liens indique les zones où les gènes sont dans le même ordre sur les homoeologues. Dans l'ensemble l'ordre est bien conservé entre homoeologues. On observe tout de même des régions où l'ordre est inversé et des régions dupliquées mais globalement, ça montre que la structure est la même entre les chromosomes homoeologues.
D'où vient la paléotétraploide ?
Adineta vaga est diploïde car il possède des paires de chromosomes homologues et il est paléotétraploide car on trouve des copies de ces chromosomes avec une plus grande divergence. Pour expliquer d'où vient la paléotétraploidie, il y a deux hypothèses. Soit l’ancêtre d‘A. vaga s’est hybridé avec une autre espèce proche. Donc l’ancêtre était sans doute sexué et a perdu la sexualité suite à cet événement d'hybridation (ce qui a déjà été observé dans d'autres espèces). Dans ce cas, on parle de chromosomes homoeologues. Soit l’ancêtre a eu un « accident génétique » et a dupliqué l’entièreté de son génome. Dans ce cas, on parle de chromosomes ohnologues. Ce phénomène a déjà été observé dans l’évolution d’énormément d’espèces (dont l’humain). Par exemple, chez les vertébrés (dont nous faisons partie) des études récentes estiment que la plupart des espèces ont connu au moins 2 duplications complètes du génome. De plus, chez certains poissons on en a documenté une troisième duplication plus récente. Quoi qu’il en soit, concernant Adineta vaga, on ne sait pas encore avec certitude quelle hypothèse est la bonne.
Transmission et mutations des séquences
Comme le génome est la seule chose qui se transmet de génération en génération (et oui le seul dénominateur commun à plusieurs générations consécutives, c’est le génome), c'est important d'étudier ce génome pour comprendre l'évolution.
Les séquences du génome ont une histoire évolutive. Elles gardent les marques des événements génétiques qui ont eu lieu dans les précédentes générations, la paléotétraploidie en est un exemple.
Les événements génétiques laissent des marques sur les séquences elles mêmes. Il existe différents types d'événements génétiques qui altèrent les séquences. On parle de mutation quand une paire de base est mal copiée et qu'on introduit une autre paire à sa place. Il y a d’autres mécanismes qui laissent des traces et pas forcément des mutations ponctuelles. Par exemple, la recombinaison permet de réparer les cassures de l’ADN d’une version d’un chromosome (par exemple la version paternelle) en utilisant l’information de l’autre version (par exemple la version maternelle). Ce processus peut effacer les mutations et rendre des copies plus identiques mais il permet aussi de créer de la diversité pendant la méiose en rejoignant des séquences du génome paternel avec des séquences du génome maternel, ces séquences ne se seraient pas rejointe autrement. Il est aussi possible d'effacer une séquence de plusieurs paires de bases, ce qu’on appelle une délétion. Et à l'inverse, il est possible d'insérer une séquence (une insertion) ou de recopier deux fois la même séquence par erreur (une duplication).
Tous ces types d'événements et d'autres dont je ne parle pas ont lieu plus ou moins fréquemment en fonction de plusieurs paramètres comme l’emplacement dans le génome, la force de la sélection sur les séquences et les gènes concernés, les propriétés cellulaires de l’organisme, le mode de reproduction de l'organisme, la consanguinité, etc.
Une vie sans sexe et qu'est ce qu'un génome peut nous apprendre ?
Comme A. vaga se reproduit sans mâles, nous avons un avantage, nous pouvons facilement détecter certains événements génétiques, notamment la recombinaison.
Dans un organisme sexué, quand les chromosomes recombinent pendant la méiose, une partie des chromosomes impliqués est devenue identique. Mais seulement la moitié du génome d’un individu est transmise dans un ovule ou un spermatozoïde, on ne sait pas dire si la copie qui reste dans le descendant est une copie originale ou une copie qui a recombiné. Pour ça on est obligé d’aller voir chez les frères et sœurs ou chez les parents, ce qui nous oblige à observer plusieurs individus et à comparer les séquences pour être sûr que la région a recombiné ou non.
Depuis 2013, on pense que A. vaga utilise une mitose pour se reproduire parce qu'on pensait que le génome était une mosaïque sans chromosomes homologues et donc qu'ils ne pouvaient pas s’apparier. Notre assemblage démontre que des chromosomes homologues existent et par conséquent il est possible qu’ils s’apparient. Le principal argument pour une reproduction par mitose est invalidé.
Cela dit, ça ne signifie pas que Adineta vaga utilise une méiose à la place. On revient à la case départ : on ne sait pas comment A. vaga produit ses oeufs.
Heureusement, on peut avoir des indices. Une des caractéristiques de la méiose est que les chromosomes s'apparient et recombinent. L'avantage de ce modèle est qu'il n'y a pas de fécondation donc en observant un génome de la génération 0 et un génome de n'importe quelle génération suivante, on devrait pouvoir dire directement si on observe de la recombinaison.
Ce dernier point n'est pas trivial donc pour bien comprendre, il faut se souvenir que les chromosomes homologues ne sont pas identiques : ils ont en moyenne 1.7% de divergence. Cela signifie que si les homologues s'apparient et recombinent une partie d'un des deux chromosomes sera "convertie" et maintenant deux séquences seront identiques. C'est montré schématiquement ci-dessous (je simplifie beaucoup sur cette figure, ce n'est pas réellement le processus de recombinaison mais il illustre bien le genre de chose qu'on peut attendre s'il y a une recombinaison).
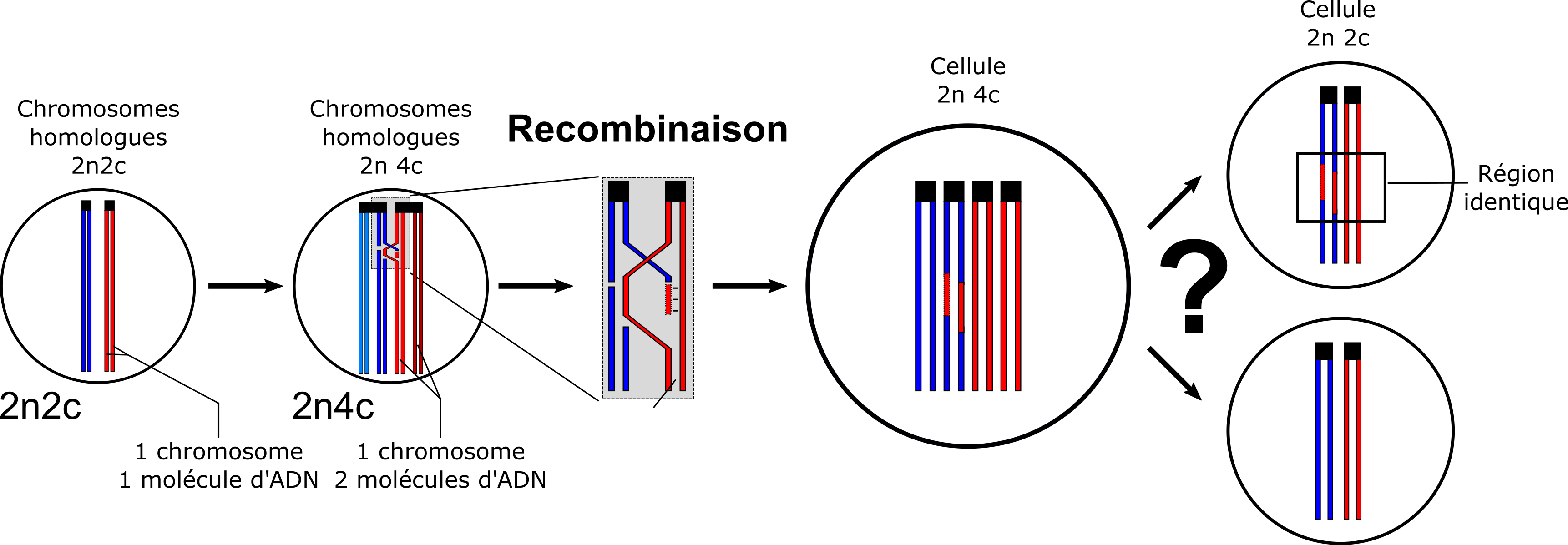
Vous remarquerez sans doute que sur ce schéma, on peut s'attendre à voir des régions identiques dans les cellules s'il y a de la recombinaison mais on peut aussi ne pas trouver de région identique malgré qu'il y a eu de la recombinaison. C'est dû au fait qu'on ignore précisément ce qu'il se passe à l'étape marquée par un "?", on appelle ça la ségrégation. Dans une méiose classique, la cellule 2n4c va d'abord séparer les chromosomes rouges des chromosomes bleus pendant la première division (méiose I) et puis dans un second temps va séparer les deux chromatides soeurs de chacun indépendamment. On se retrouverait avec 4 cellules 1n1c.
On sait que A. vaga ne fait pas de méiose classique mais on ne sait pas s'il y a de la recombinaison et s'il y en a est ce qu'elle a lieu comme montré ci-dessus ou autrement ? Et si elle a lieu comment est-ce que A. vaga fait la dernière étape de "ségrégation" ?
Pour commencer à répondre à toutes ces questions, on a cherché des régions identiques entre les chromosomes homologues. Pour faire ça, on a utilisé la même approche que pour compter la divergence entre les homologues, sauf qu'au lieu de simplement compter les endroits différents, on a regardé la taille des régions identiques entre les endroits différents.
On a deux hypothèses : soit il y a recombinaison et les régions identiques sont parfois dues à celle-ci. Soit il n'y a jamais de recombinaison et les régions identiques sont simplement liées au fait que les mutations se répartissent au hasard sur le chromosome. Avec ces deux hypothèses, on peut faire des prédictions : si les régions identiques sont uniquement le produit de la répartition au hasard des mutations ponctuelles, on peut essayer de simuler une paire de chromosomes identiques qui subiraient des mutations au hasard puis on pourrait regarder dans cette simulation la distribution attendue de la taille des régions identiques.
Mais attention, il faut tenir compte d'une chose, la sélection, (encore et toujours). La divergence n'est pas répartie de façon homogène sur le génome parce que certaines régions sont plus fortement sélectionnées que d'autres (les gènes!). Comme les gènes sont moins divergents que les autres régions on s'attend à trouver des régions identiques plus longues qu'attendues par répartition au hasard des mutations donc on va voir un signal de recombinaison alors qu'en fait on a observé juste de la sélection.
Pas de problème, comme on sait que les gènes sont là, il nous suffit d'en tenir compte : on va prendre les chromosomes de Adineta vaga et on va répartir au hasard toutes les mutations qu'on a trouvé sauf celles qui se situent dans les gènes. Ensuite, on va regarder la taille des régions identiques et on va voir si la distribution observée est différente que celle simulée avec une répartition indépendante des mutations entre les homologues.
Qu'est qu'on s'attend à voir : s'il y a de la recombinaison on devrait voir des régions identiques plus longues que dans la simulation et aussi des régions plus courtes car ça signifie qu'en réalité les mutations sont réparties avec une densité inégale à cause de la recombinaison alors que dans notre simulation la densité devrait être relativement homogène (sauf dans les gènes évidemment). Et s'il n'y a pas de recombinaison alors la simulation devrait avoir la même distribution que les résultats observés. La figure ci-dessous montre la simulation en orange et les données observées en bleu.
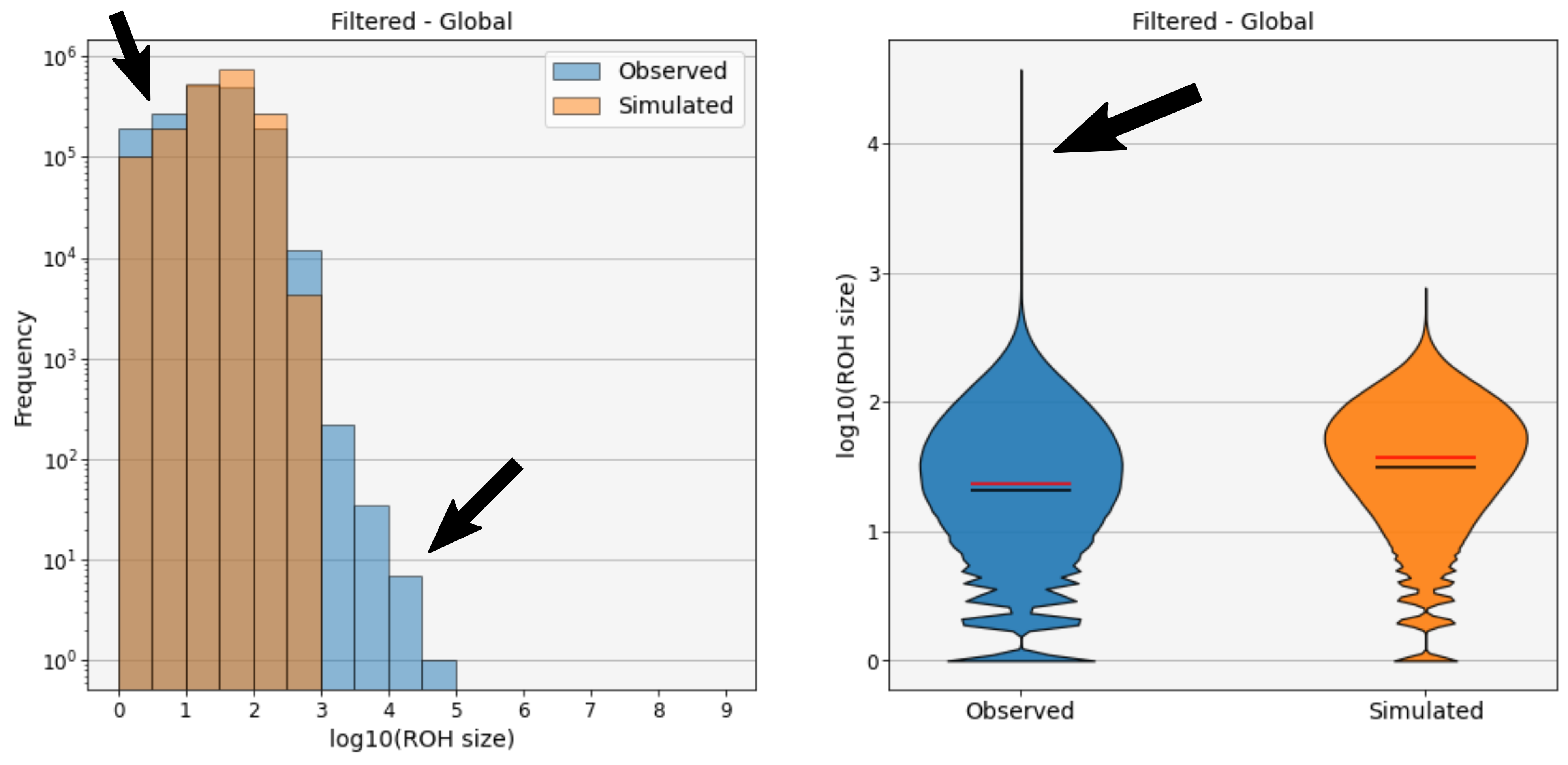
Bingo, on trouve des régions identiques plus longues que dans la simulation ! Une bonne piste pour dire qu'il y a de la recombinaison.
Est-ce qu'on peut faire encore plus fort ?
Ok, on pourrait se dire : ce n'est pas très convaincant, peut-être qu'ils n'ont pas vu certains gènes ou d'autres régions sous sélection et que ce sont ces régions qui provoquent des tailles identiques plus grande que prévu...
Heureusement, on peut faire encore plus convaincant. Quand on a discuté des premiers résultats d'assemblage avec les collègues, on s'est rapidement dit : en fait on a déjà des séquences qui proviennent d'une autre génération. Et oui, les séquences génomiques publiées en 2013 sont toujours accessibles en ligne et on pourrait les comparer avec les séquences récentes pour voir s'il y a des changements.
Donc c'est ce qu'on a fait, on a regardé dans 3 jeu de données qui datent respectivement de 2009, 2015 et 2017. Ces jeux de données ont tous le même ancêtre car ils sont tous obtenus sur des individus qui sont issu de la même souche de labo mais évidemment ceux de 2015 et 2017 sont resté 6 et 8 ans de plus dans le labo. Ils ont eu beaucoup de temps pour faire de la recombinaison!
Qu'est ce qu'on s'attend en cas de recombinaison dans un de ces échantillons ? Et bien on espère trouver des régions où il y a des mutations entre les homologues dans un des échantillons mais où il n'y en a pas dans les autres échantillons. Ca signifie que les mutations ont été effacées par la recombinaison entre les homologues.
Pour ça il suffit de reprendre à nouveau l'approche où on compte les différences dans les séquences alignées et on va faire ça pour les 3 échantillons en parallèle. Quand on trouve une région où un des trois échantillons n'a pas de différence par rapport aux autres : on a probablement trouver des régions qui ont recombiné. Afin de représenter ça de façon digeste (il y a quand même environ 1 à 2 millions de mutations au total), on ne va pas représenter directement toutes ces divergences entre les homologues, on va compter le nombre de divergence par fenêtre le long des chromosomes. S'il y a 100 divergences dans une fenêtre pour l'échantillon 2009 et qu'il y a 0 divergences dans la même fenêtre dans l'échantillon 2015 ou dans celui de 2017 alors c'est gagné : on a directement observé de la recombinaison.
Vous pouvez voir ce que ce comptage donne sur la figure ci-dessous. Les zones colorées indiquent les régions où le comptage des divergences dans chaque fenêtre est différent entre les échantillons.
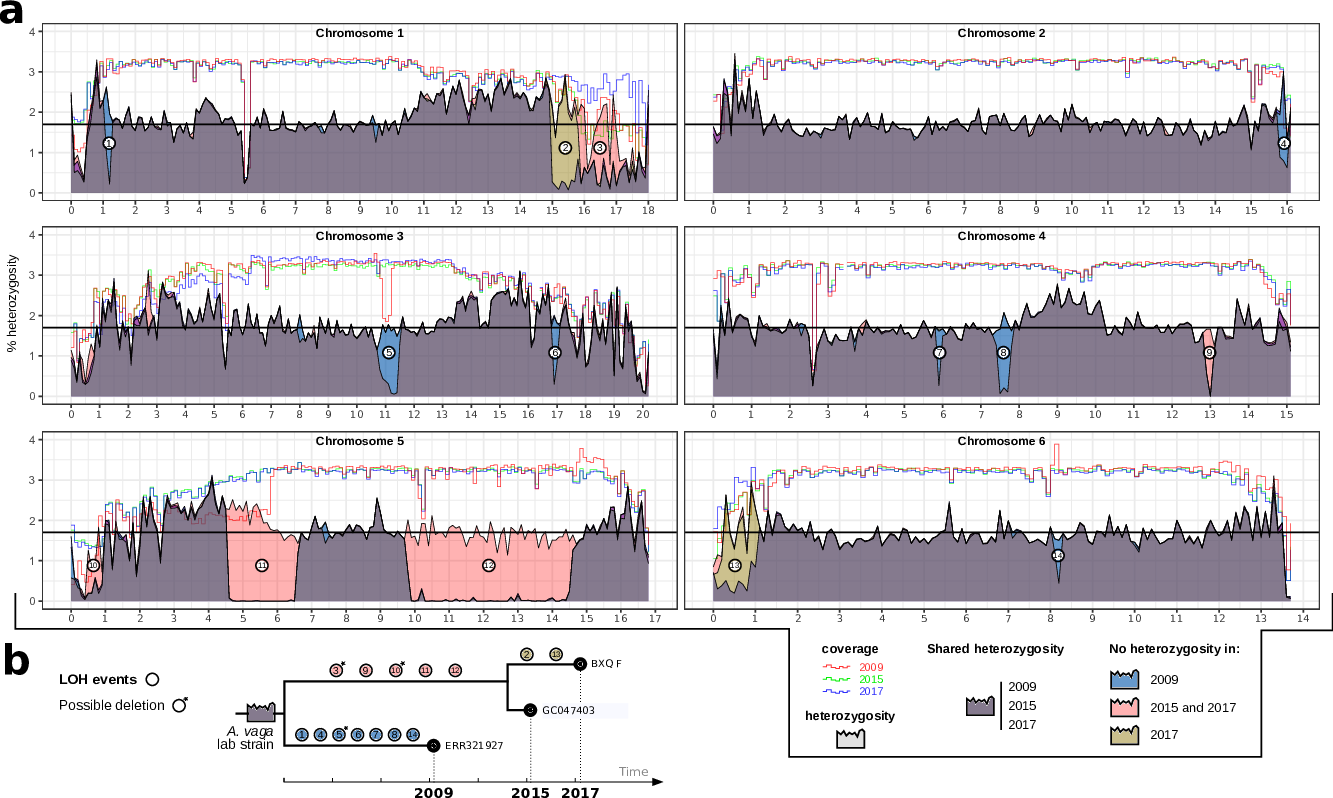
Et oui ! D’ailleurs ces régions recombinantes sont très grandes. Ces différences ne peuvent être expliquée que par de la recombinaison.
En conclusion
Nous avons démontré qu’il y a bien des chromosomes homologues dans Adineta vaga et que ces chromosomes sont capables de recombiner. Ces deux faits ouvrent de nouveau pas mal de perspectives chez les rotifères bdelloïdes. On change complètement le paradigme de 2013 et en un sens on pose plus de nouvelles questions : quel est le mode de reproduction des rotifères bdelloïdes ? Est ce qu'ils utilisent la recombinaison seulement pour réparer leur ADN ou pour se reproduire également ? Est ce que parfois ils font quand même du sexe ?
Bref, autant de questions que de mystères qu'il nous reste à élucider. Cela dit, on avance bien et on avance vite, j'ai bon espoir que ces questions restent sans réponses pendant seulement quelques mois ou quelques années avant qu'on passe encore aux suivantes.
Source principale :
Simion, P., Narayan, J., Houtain, A., Derzelle, A., Baudry, L., Nicolas, E., Arora, R., Cariou, M., Cruaud, C., Gaudray, F. R., Gilbert, C., Guiglielmoni, N., Hespeels, B., Kozlowski, D. K. L., Labadie, K., Limasset, A., Llirós, M., Marbouty, M., Terwagne, M., … Doninck, K. V. (2021). Chromosome-level genome assembly reveals homologous chromosomes and recombination in asexual rotifer Adineta vaga. Science Advances, 7(41), eabg4216. https://doi.org/10.1126/sciadv.abg4216
Accès en ligne : https://www.science.org/doi/10.1126/sciadv.abg4216
Comments powered by CComment